RESEARCH FOCUSS IN KRANIAS Laboratory
Calcium Homeostasis and Cardiac Muscle Contractility
Heart failure is the single leading cause of death and the only disease that is increasing in incidence and prevalence with an overall five-year mortality rate of 50%. Current pharmacological treatment of this devastating condition focuses on symptoms rather than the underlying cellular mechanisms responsible for weakness of the heart muscle. Cellular hallmarks of cardiac failure include marked ventricular hypertrophy or dilation and abnormal cytosolic calcium handling, associated with impaired contractility. The overall goal of our research program is to elucidate the regulatory mechanisms and signaling pathways underlying calcium homeostasis in cardiac muscle and the alterations in these pathways underlying the heart failure phenotype. Our current approach is to use Molecular Genetics and generate mouse models, with alterations in the expression levels of key Ca-handling proteins to determine their physiological and pathophysiological significance in vivo. These genetically altered models are characterized at the molecular, biochemical and physiological levels, using state of the art techniques, including Genomics, Proteomics and Bioinformatics. Our basic studies are also extended to the Clinical arena to determine whether genetic variants in the key calcium cycling proteins may serve as prognostic or diagnostic markers for heart disease or even as therapeutic targets.
The Sarcoplasmic Reticulum (SR)
The sarcoplasmic reticulum (SR) is an internal membrane system in muscle and it has been implicated as a major contributor to the depressed function in heart failure. The SR acts as a calcium source during contraction and a calcium sink during relaxation. Relaxation is mediated by the transport of calcium into the sarcoplasmic reticulum lumen by a Ca-ATPase, which is under reversible regulation by phospholamban, a low molecular weight phosphoprotein. Calcium then binds to calsequestrin in the lumen of the SR. For the initiation of contraction, calcium is released through the calcium channels or ryanodine receptors, which are under regulation by junctin, triadin and the histidine rich calcium-binding protein. Thus, the SR is the major regulator of Ca2+-handling and contractility in muscle (See Figure Below).
However, our original and simple understanding of SR Ca-cycling has been evolving through the years and our studies have identified a multimeric protein complex that regulates SR Ca-transport composed of SERCA, Phospholamban (PLN), the anti-apoptotic protein HAX-1, Protein Phosphatase 1 with its regulators Inhibitor-1 and Hsp20 and the Histidine Rich Ca-Binding Protein. We call this interactome as the SERCA/PLN Regulatome (see Figure below).
SR Ca-Regulatome in Cardiac Function
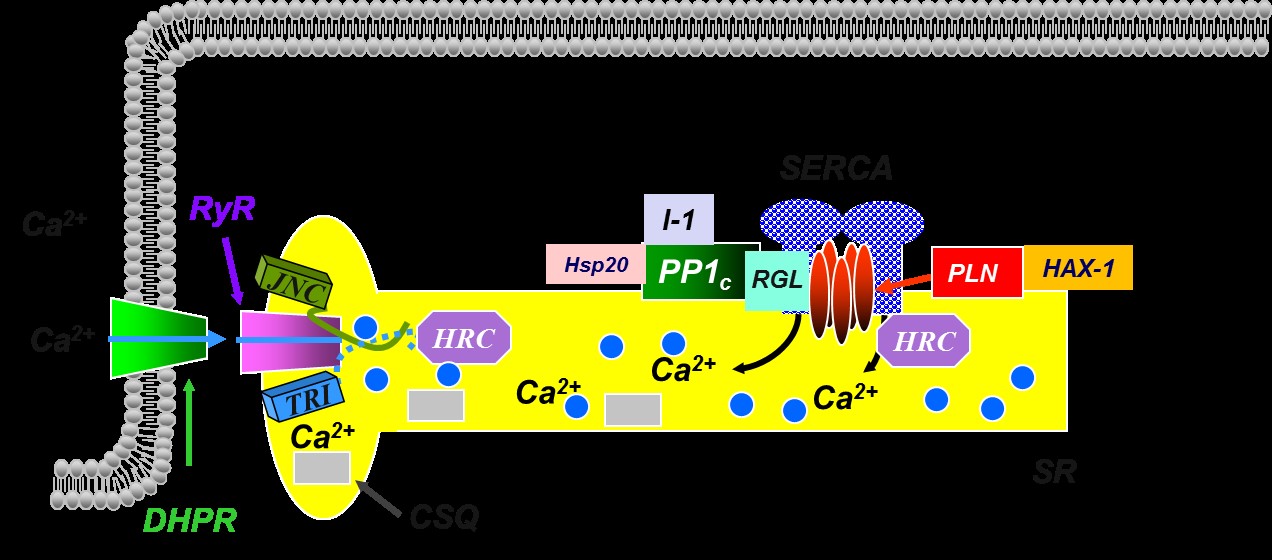
To elucidate the physiological significance of the key SR/Ca-regulatory proteins in vivo, we use Molecular Approaches with emphasis on generation of mouse models with alterations in the expression levels or the activity of each of these proteins. Our models are characterized at the Molecular, Biochemical and Physiological levels, using state of the art techniques, including Functional Genomics and Proteomics. This integrative approach reveals unique phenotypes, which provide significant insights into the key regulatory players in muscle and their role in health and disease. Furthermore, studies on: a) muscle development; b) aging; and c) development of hypertrophy with its transition to heart failure, or various myopathies in the mouse models, offer unique insights into the role of each of these calcium-handling proteins in cardiac myocyte contractility and survival. Identification of compensatory mechanisms, accompanying each gene alteration, also provides an in-depth understanding of the molecular communication pathways in cardiac muscle. Our goal is to build a comprehensive understanding of the sarcoplasmic reticulum role in calcium handling mechanisms that impact on the control of muscle function and survival/death in health and disease.
Phospholamban
Phospholamban (PLN) is a 52 amino acid protein of 6.1 kDa that forms a homopentamer. In the dephosphorylated state, it is an inhibitor of SERCA2a activity, but inhibition is relieved upon phosphorylation of PLN. In vitro studies indicated that PLN can be phosphorylated at three distinct sites by various protein kinases: serine 10 (Ser10), by protein kinase C; serine 16 (Ser16), by cAMP- or cGMP-dependent protein kinase; and threonine 17 (Thr17), by Ca2+-calmodulin-dependent protein kinase. Phosphorylation of PLN is associated with stimulation of the initial rates of cardiac sarcoplasmic reticulum Ca2+ uptake at low Ca2+, resulting in an overall increase in the affinity of the SERCA2a for Ca2+.
The role of PLN in the regulation of basal contractility has been elucidated through the development of genetically engineered mouse models. Characterization of the cardiac phenotype of our PLB-knockout mice indicated that PLN is: a) a key determinant of the affinity of the SR Ca2+-ATPase for Ca2+; and b) a major regulator of calcium homeostasis and contractility in cardiac, smooth and slow-twitch skeletal muscles. PLN is also a key mediator of the b-adrenergic responses in the heart. Furthermore, using transgenic technology, we generated mice with cardiac-specific or fast-twitch skeletal muscle-specific overexpression of PLN. Characterization of these models indicated that: a) only a fraction of the cardiac sarcoplasmic reticulum calcium-pumps is under regulation by PLN in vivo, suggesting that this may constitute an important "brake mechanism" in the heart; and b) ectopic expression of PLN in fast-twitch skeletal muscles results in depressed Ca2+-transients and contractility (See Figure Below). These models with altered Ca2+ homeostasis and contractility have been valuable tools for our research group and other laboratories around the nation to test specific hypotheses and further define the mechanisms regulating excitation-contraction coupling in muscle.
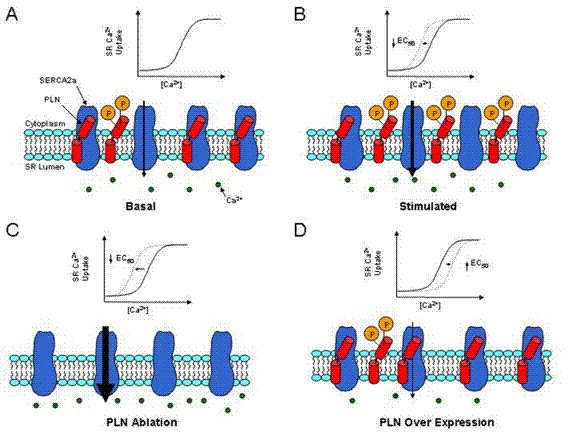
Schematic representation of the effects of PLN ablation and overexpression in the heart.
HAX-1
HAX-1 was originally identified to interact with HS1, a protein with proposed involvement in B cell signal transduction. Recently, we identified HAX-1 as a PLN-binding partner. With the use of genetic over-expression and ablation techniques, we generated mouse models which revealed that HAX-1 overexpression enhances the inhibitory effect of PLN on SERCA, leading to slower SR calcium kinetics, while HAX-1 ablation has the opposite effects. Thus, SR Ca-transport is regulated by the HAX-1/PLN/SERCA multimeric complex. Moreover, this complex plays a critical role in controlling the activation of endoplasmic reticulum stress induced apoptosis, which impacts cell survival during ischemia/reperfusion injury. Current studies in our lab are focusing in dissecting the contribution of HAX-1 to ER/SR function, using our genetic models including a cardiac specific inducible HAX-1 gene ablation mouse. We are also interested in understanding how the HAX-1/PLN/SERCA complex mediates the crosstalk between calcium homeostasis and cell death responses at the ER/SR membrane.
HAX-1 also localizes to the mitochondria in cardiac cells, which may regulate its normal function, such as cell-death activation and energy production. Since the role of HAX-1 in cardiac mitochondrial function has not been elucidated yet, our lab seeks to understand how mitochondrial HAX-1 impacts apoptosis/necrosis activation and energetic function, which may dictate the manifestation and progression of various cardiomyopathies. As diseased hearts demonstrate both ER/SR and mitochondria dysfunction, dissecting the role of HAX-1 in both organelles will provide significant insights into its role in cardiac physiology and pathophysiology.
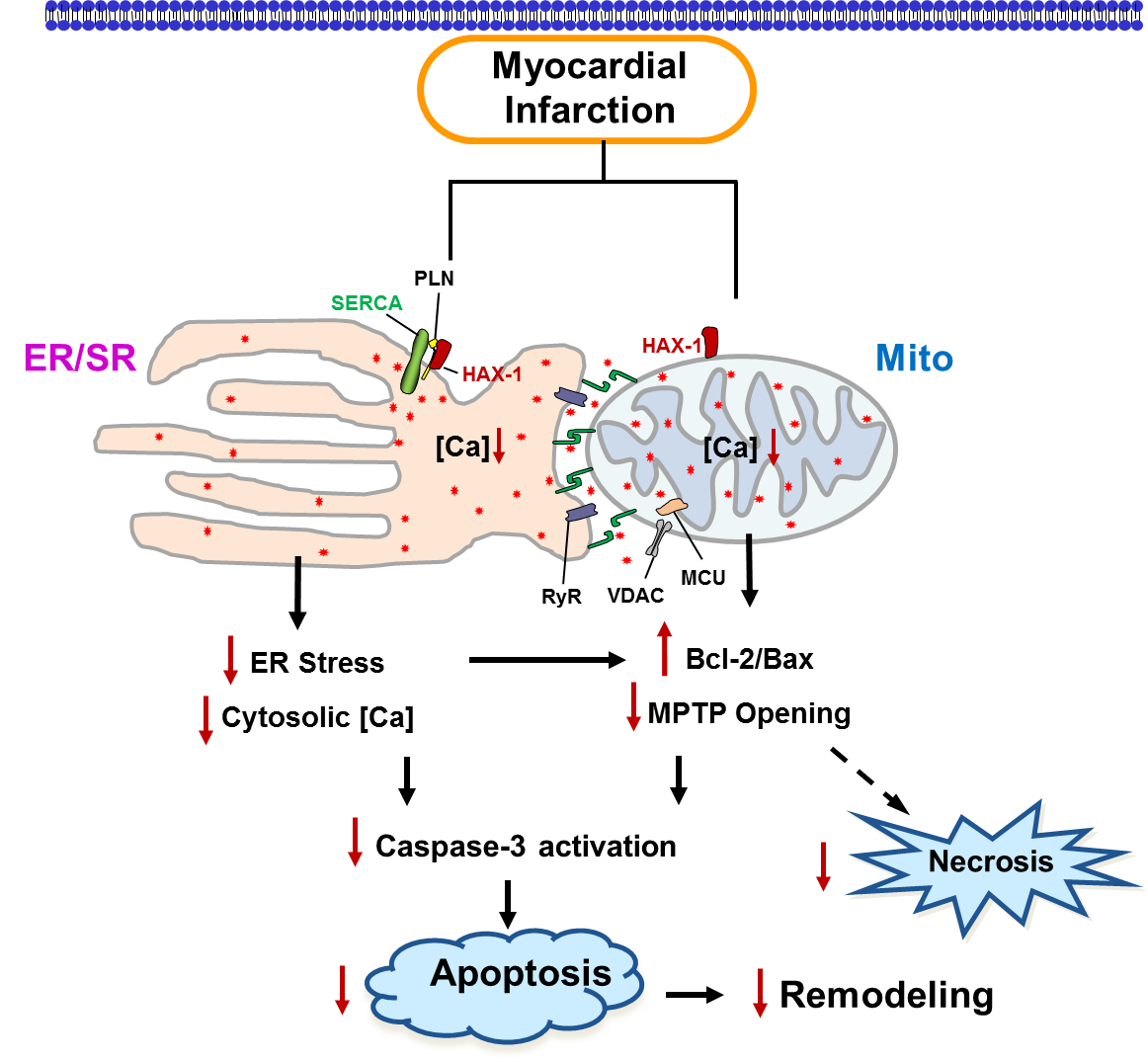
Schematic of the HAX-1 regulatory effects on survival of cardiac cells
Protein Phosphatase-1 and Inhibitor-1
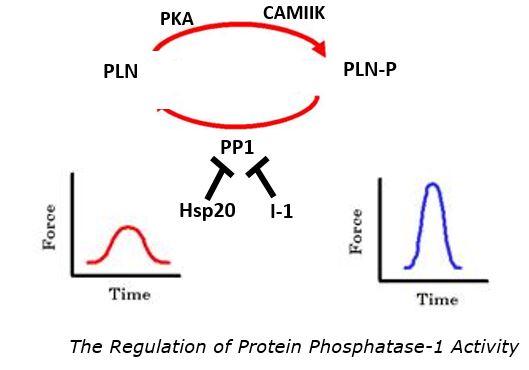
Abnormal calcium cycling, a characteristic of experimental and human heart failure, is associated with impaired sarcoplasmic reticulum calcium uptake activity, as described above. Protein phosphatase 1 (PP1) is responsible for the dephosphorylation of PLN, resulting in depressed calcium cycling and decreased contractile force in the heart. In heart failure, increased PP1 activity coupled with decreases in cAMP-pathway signaling contributes to decreased cardiac function (Figure below).

Figure: The Balance of Protein Kinases and Phosphatases
The increased PP1 activity in heart failure is partially due to dephosphorylation and inactivation of its Inhibitor-1 (I-1), promoting dephosphorylation of phospholamban and inhibition of the sarcoplasmic reticulum calcium-pump. Indeed, cardiac-specific expression of a constitutively active Inhibitor-1 (I-1c) results in selective enhancement of phospholamban phosphorylation and augmented cardiac contractility at the cellular and intact animal levels through inhibition of Protein Phosphatase-1. Furthermore, the ß-adrenergic response is enhanced in the transgenic hearts compared with wild types. On aortic constriction, the hypercontractile cardiac function is maintained, hypertrophy is attenuated and there is no decompensation in the transgenics compared with wild-type controls. These studies in cardiac protection were extended to test the hypothesis that I-1 overexpression in the heart could restore cardiac function in failing hearts. Indeed adenoviral or AAV-mediated gene delivery of the active inhibitor-1 greatly restores function and partially reverses remodeling in the setting of pre-existing heart failure. These studies are done in collaboration with Dr. Roger Hajjar's lab currently at Mt Sinai Medical center and are currently in clinical trials.
In addition, our lab has recently discovered a novel regulator of PP1 activity in addition to I-1, the heat shock protein 20 (Hsp20) (Fig 2). Hsp20 has been shown to play a cardioprotective role in the heart under stress conditions, and it has also been identified as a regulator of calcium-cycling through direct interaction with PP1, associated with PLN. Inhibition of PP1 activity increases the phosphorylation of PLN, thereby enhancing cardiac function. Current studies in our lab are aiming to understand the dual regulation of PP1 by Inhibitor-1 and Hsp20.
Histidine-Rich Calcium Binding Protein and the S96A Human Variant
The histidine-rich Ca binding protein (HRC), a 165 kD SR protein, has recently emerged as a regulator of Ca cycling during excitation-contraction coupling. HRC regulates the SR Ca uptake and release by direct interaction with the SR Ca ATPase (SERCA) and with Triadin, a member of Ca release complex. The functional importance of HRC is underscored in our recent identification of the human genetic variant (Ser96Ala) that is linked to life threatening ventricular arrhythmias and sudden death in a well‐characterized cohort of patients with idiopathic dilated cardiomyopathy (DCM). The S96A variant exhibits a strong dosage effect in ventricular arrhythmia as reflected in the findings that Ala/Ala homozygous individuals exhibit an increased risk for sudden cardiac death as compared to the Ser/Ala heterozygotes. To investigate the in vivo pathophysiological mechanisms associated with the S96A variant, we generated mice with the human S96 and A96 HRC in the heart of a HRC null mouse (HRC-KO) background to assess the function in the absence of the endogenous protein. Our findings from in vitro and in vivo studies revealed that the underlying cause for the increased risk of sudden cardiac death in the DCM patients with the A96 variant includes: a) increased SERCA2a inhibition and reduced SR Ca-load; b) enhanced Ca-leak through the ryanodine receptor (RyR); and; c) aberrant Ca handling in the myocytes that manifested in spontaneous Ca-waves and arrhythmogenic delayed after depolarizations.
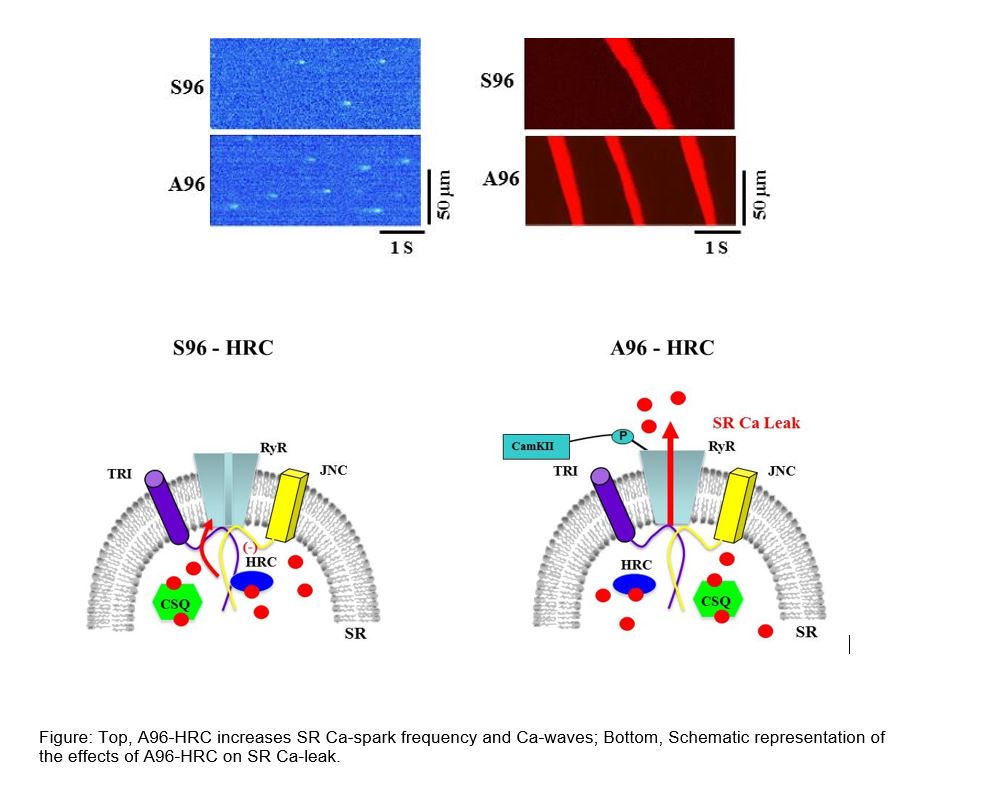
SR Calcium Cycling Protein Genetic Variants in Human Heart Failure
Our basic studies have recently been extended to the Clinical Arena, in collaboration with our Cardiology Colleagues. We have examined the expression levels of the sarcoplasmic reticulum (SR) Ca-handling proteins in normal and failing human hearts, since their depressed cardiac function has been suggested to reflect abnormalities at the SR level. Our findings indicate that the levels of SR Ca-cycling proteins, such as phospholamban (PLN) relative to the SR Ca-ATPase (SERCA2a) are increased in human heart failure. This increase may constitute a contributing factor to diastolic dysfunction and depressed contractility exhibited by the failing heart, as indicated by our studies in the transgenic mice. Furthermore, decreased phosphorylation of PLN, without alterations in its protein levels, has been suggested to contribute to the depressed function in human heart failure. The attenuated PLN phosphorylation is at least partially due to increased protein phosphatase-1 activity, reflecting inactivation of its Inhibitor-1 protein in human failing hearts. Indeed, both Inhibitor-1 and PLN phosphorylation are decreased in human heart failure. Thus, our hypothesis is that: a) the activities of SERCA2a, PLN and Inhibitor-1 are major determinants of cardiac function; and b) naturally occurring mutations in the SERCA2a, PLN and Inhibitor-1 genes may alter SR Ca-cycling and contractility, leading to remodeling and heart failure. In our lab, we are examining whether naturally occurring genetic variations (polymorphisms) may exist in the coding regions of these major SR Ca-handling proteins which may act as "modifiers" of the clinical time course of the disease in patients with heart failure. These studies are done in collaboration with the Cardiologists at several Institutions.
Human SERCA2a Mutations
We screened a large number of dilated cardiomyopathy patients and identified some mutations in the SERCA2a gene. However, none of these variants changed the amino acid composition, indicating that this gene might be evolutionary conserved in humans.
Human Phospholamban Mutations
In pursuit of identifying naturally occurring mutation(s) in the human phospholamban (PLN) gene, a truncation mutation (L39stop) was identified in individuals from two families with inherited dilated cardiomyopathy that resulted in dramatically diminished myocardial PLN protein content, consequent loss of PLN inhibition of SR Ca2+-ATPase, and the development of heart failure and early mortality in homozygous individuals (Figure below). These findings demonstrate that, in contrast to mice in which PLN-deficiency enhances myocardial inotropy and lusitropy without adverse effects, PLN is essential for cardiac health in humans and its absence results in lethal heart failure. In addition, a second mutation in the coding region resulted in deletion of amino acid 14 in PLN (R14del). The heterozygous carriers developed left ventricular dilation, dysfunction, episodic ventricular arrhythmias and death by mid-age. Overexpression of R14del-PLN in the mouse resulted in super-inhibition of SERCA2a, early cardiac pathology and death. The functional significance of this mutation is now studied by CUREPLaN, a consortium that is funded by the Leducq Foundation.
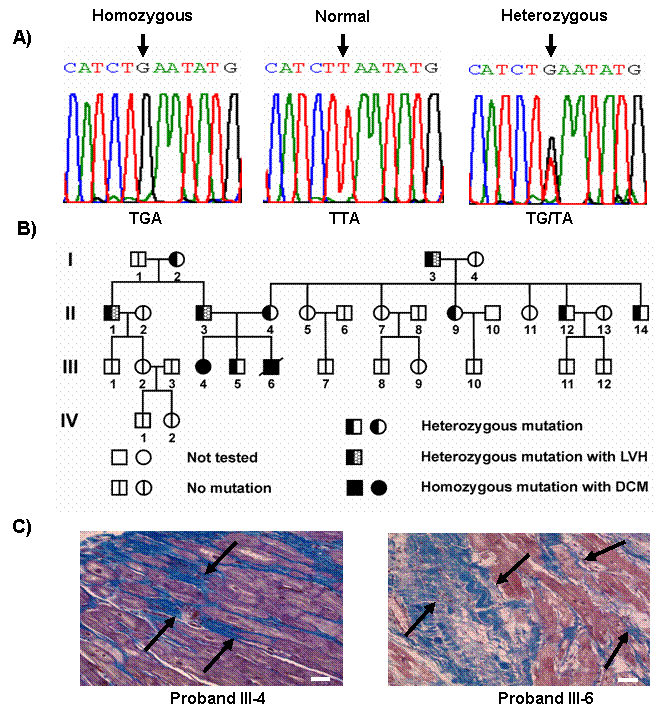
Human Inhibitor-1 Mutations
Parallel studies on the Inhibitor-1 of protein phosphatase-1, which is currently in clinical trials, identified genetic variants that alter the function of Inhibitor-1 and may modify the time course of heart failure development.
Contact Us
Department of Pharmacology, Physiology,
and Neurobiology
College of Medicine
PO Box 670576
Cincinnati, OH 45267-0576
Ms. Jennifer Bedel
Associate To The Chair
Phone: 513-558-5636
Email: bedelj@ucmail.uc.edu